Translate this page into:
Beta thalassaemia trait - fitness for fighter flying: Two case reports
Abstract
Two cases of highly experienced fighter pilots in whom thalassaemia trait was detected, are reported. During routine medical evaluation they were detected to have anemia, which was followed by hemoglobin electrophoresis study, and the diagnosis of thalassaemia trait was clinched. They were subjected to aero medical evaluation at Institute of Aerospace Medicine (IAM) by exposure to simulated flight conditions of hypoxia in an altitude chamber. They were also evaluated for the tolerance of acceleration stress in the human centrifuge. No red cell damage was demonstrated during repeated and prolonged exposure to altitude of 4,570 m (15,000ft). Clinical hypoxia induced by disconnecting oxygen supply at 9,140 m (30,000ft) also did not produce any evidence of untoward symptoms/signs or red cell damage. The pilots were assessed to be fit for full flying duties on fighters. It is well known that thalassaemias produce a low grade anemia that can cause problem at high altitude, however no such effects could be demonstrated in these two cases. Thalassaemia cases are very rare in modern aviation, hence a reconsideration and introduction of new policy is stressed.
Keywords
Beta thalassaemia trait
Anemia
Hb electrophoresis
Aviation stresses

The thalassaemias probably constitute the world's largest gene disorder. Beta thalassaemia occurs widely in a belt extending from southeast Asia, through India, the Middle East, the Mediterranean (as far North as Romania and Yugoslavia), and to North and West Africa. Carrier frequencies can vary from 2 to 30% in these populations [1].
Beta thalassaemia also occurs sporadically in every racial group. Thalassaemias produce a low-grade anemia that can cause problems at altitude. Splenic enlargement and worsening of the anemia can occur under conditions of stress. Splenectomy results in a greater risk of overwhelming infection and of severe malaria, which can affect an aviator's fitness to fly [1]. Hypochromia and microcytosis due to reduced amount of hemoglobin characterize all forms of Beta thalassaemia. In heterozygote’s (Beta thalassaemia trait) this is the only abnormality seen. Most patients with thalassaemia trait have no symptoms, live normal life spans and do not require any treatment [2]. Homozygous Beta thalassaemia or deletions in more than two of the Alfa chains are almost always severely symptomatic or anemic, and as such rarely make it into the military. Also, in these cases clinical severity precludes flying. Bone marrow transplant in childhood is the only curative therapy. [2] This paper reports two cases of Beta thalassaemia trait diagnosed in two very highly experienced fighter pilots of Indian Air Force. These patients were referred to IAM, IAF for evaluation.
Case Study
Case-I
MPS, a 39 yrs old fighter pilot in IAF completed 2000 hrs of flying in different aircraft with 1450 hrs on type was diagnosed as a case of 'NIDDM' in Sep 96 later cleared, He was detected to have ECG abnormality in Mar 97 for which he was investigated and cleared. Later in Jan 99 he was found to have 'Bilateral Sensor neural Hearing Loss' for which he was put on restricted flying category. During his review in Jan 99 he was detected to have 'Hypo chromic anemia' which on electrophoresis was found to be Beta Thalassaemia minor (Fig 1). He was awarded a non-flying medical category A4G2 for 12 weeks. After 12 weeks he was reviewed at AFCME in May 99 and continued in A4G2 for another 4 weeks. Subsequently in Jun 99 he was again reviewed in AFCME and was upgraded to a restricted flying category A2G2 for 12 weeks. He was declared ‘fit to fly transport and helicopters. In Oct 99 he was reviewed at AFCME and placed in permanent restricted flying category A2G2; fit to fly transport and helicopters only. However after his annual review at AFCME while approving the Medical Board proceedings, Approving Authority referred him to IAM, Bangalore for evaluation under aviation stresses to consider his suitability for flying fighter aircraft. The pilot was reviewed at IAM in Feb 2001.
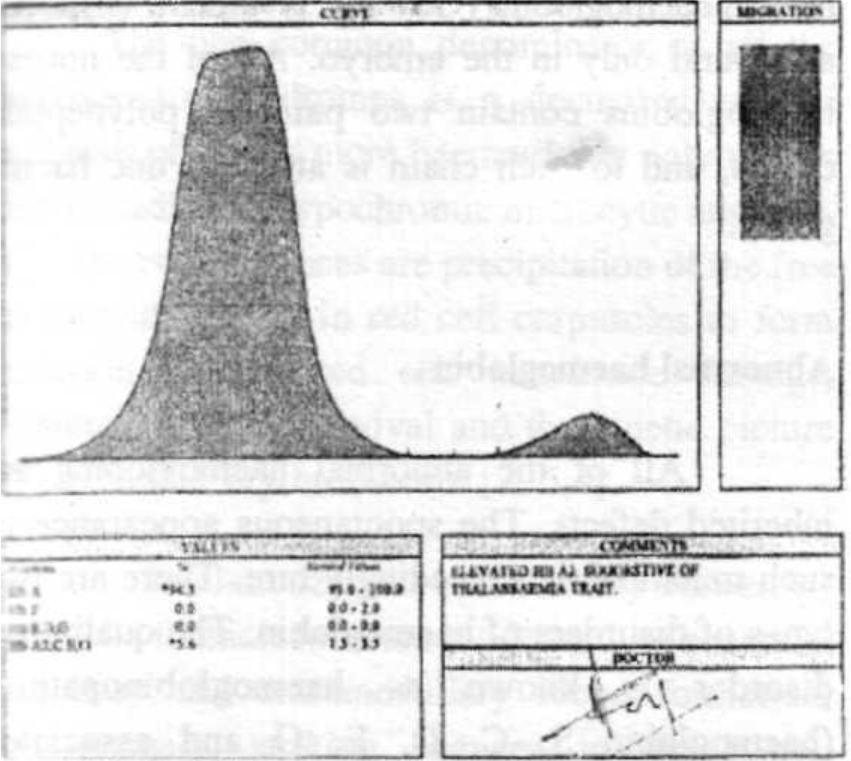
- Electrophoresis of Subject No 1
Case-II
SBC, a 41 yrs old fighter pilot in IAF, who had completed 3000 hrs of flying in different aircraft with 2000 hrs on type, was diagnosed as a case of ECG abnormality during AME in Apr 92 and followed up in low medical category A4G3 (T-12+12 weeks) and upgraded to A2G1 (Perm) in Jul 92. In Sep 95 during annual review at IAM, he was detected to have hypo chromic, microcytic anemia (Hb-10gm/ dl). Medical category was not lowered and he continued in A2G1. In Sep 96 and Oct 97 during annual medical review Hb was found to be 14.5 gm/ dl and 13.8 gm/dl respectively. In Sep 98 he was detected to have hypo chromic, microcytic anemia. He was put in LMC A4G3 (T-8 weeks) and advised oral haematinics. In Dec 98 at PGI Chandigarh Hemoglobin electrophoresis revealed raised HbA2 (level was not specified). Total iron content and iron binding capacity was found to be within normal limits. No evidence of sickle cell trait or any other haemoglobinopathy was detected. He was labeled as a case of Beta Thalassaemia Minor (trait) and placed in category A3G3 (T-12 weeks) and made unfit for fighter flying by Approving Authority. In Apr 99 he was reviewed at AFCME and recommended A2G2 (Perm), 'Fit' for transport/helicopter. Approving Authority did not concur a permanent category but advised him to be reviewed after 12 weeks at IAM along with hypoxia and acceleration stresses. In Jul 99 the pilot was reviewed at IAM and subjected to hypoxia and centrifuge runs. His responses were found to be within normal limits. He was R recommended med cat A2G2 (Perm), fit to fly fighters. ever, Approving Authority again modified the recommendation and made him fit to fly transport and helicopters only. In Jul 2000 he was reviewed at AFCME and continued in A2G2 (P), fit for transport/ helicopters.
Approving Authority in Jul 2001 directed the individual for fresh evaluation with repeat h\poxia and centrifuge runs at IAM. The pilot was then reviewed at IAM on 30 Jul 2001.
Past Hb profile
The Hb profile of Subject No 1 and Subject No 2 is given in Tab 1 and Tab 2 respectively Investigations Both the pilots were subjected to a thorough general physical and systemic examination. The clinical investigations included electrocardiography, X-ray chest PA and ultrasonography of abdomen. Hematological investigations like complete haemogram, RBC fragility, ESR, RBC count, WBC count and peripheral blood film examination were also carried out (Table 3). Biochemical investigations included lipid profile, urea, creatinine and standard glucose tolerance test. Total serum iron content and iron binding capacity was done and found within normal limits. Hemoglobin electrophoresis study (Fig 1 and Fig 2) was done for the qualitative and quantitative analysis (Table 4). Urine was examined. Both were subjected to aviation stresses in the form of altitude chamber run and human centrifuge evaluation.
| Jun 82 | Initial Entry | 15.0 gm% |
| Sep 96 | NTDDM | 14.2 gm% |
| Sep 97 | Review CME | 11.5gm%# |
| Mar 98 | Review CME | 12.8 gm% |
| Jul 98 | Review CME | 13.0gm% |
| Nov 98 | Review CME | 10.5 gm% |
| Jan 99 | Review CME | 11.5 gm% |
# Given a course of Hetrazan
| Jun 80 | Initial Entry | 15.5 gm% |
| Apr 92 | PRBBB | 11.2gm% |
| Oct 94 | Annual Med | 11.0gm% |
| Sep 95 | Review | 10.9 gm%* |
| Sep 96 | Review | 14.5 gm% |
| Oct 97 | Review | 13.8 gm% |
| Sep 98 | Review | 11.5 gm%## |
Dec 98 brought under care of Hematologist
# PC V 37, Microcytic, Hypo chromic Anemia: Haematinics at Base
# # Cat Lowered for Anemia
| Parameters | Case-I | Case-II |
|---|---|---|
| Hb (gm %) | 13.4 | 11.8 |
| ESR(mmls,hr) | 09 | 10 |
| TRBqmfl/cmm) | 5.0 | 4.4 |
| TWBC (/cmm) | 7,900 | 6,200 |
| DLC (%) | P58,E04,L36, | P59305L33, |
| M02300 | M03,BO0 | |
| PCV (%) | 42 | 37 |
| MCV(fL) | 84 | 84 |
| MCH (pg) | 27 | 27. . .. |
| MCHC (%) | 31.8 | 32 |
| Peripheral Blood Smear | Majority of red cells are »ormo cytic & normo- chromic, few microcytic and hypochrc seen. No other ■ abnormal cells seen | Microcytic, hypo chromic anemia. No abnormal cells found |
| Parameters | Case-I | Case-II |
|---|---|---|
| Hb(gm%)(13- 15) | 13.4 | 11.8 |
| Hb genotype | Not done | Not done |
| HbA% (96-98.5) | 94.3 | 93.21 |
| HbA2% (1.5- 3.5) | 5.7 | 6.79 |
| HbF%40-2.0) | 0.0 | 0.0 |
| HbS% (0.0) | 0.0 | 0.0 |
| HfoCAG, Lepore (%) | 0.0 | 0.0 |
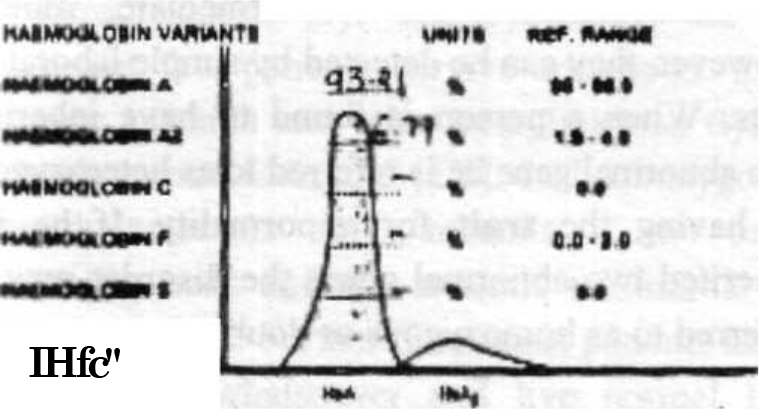
- Electrophoresis of Subject No 2
DiscHSsioa
The Iron cycle
Iron (Fe) travels from plasma to the marrow where it is incorporated into hemoglobin. It is then released with the mature red blood cells (RBCs) into the circulation,-%.-, .r: Fe span of about 120 days, the red cell is engulfed by cells of the reticuloendothelial system (EES). Here, the iron is extracted from hemoglobin and returned to plasma where it becomes bound to transferring, completing the cycle.
Globin
From the standpoint of protein structure, there are several normal hemoglobin’s.
Hemoglobin A (alpha2Abeta2A) or adult hemoglobin comprises 96 to 98% of the Hemoglobin in adults. Most of the remainder is structurally different hemoglobin called hemoglobin A2 (alpha2Adelta2A). In foetal life and early infancy the predominant hemoglobin is foetal hemoglobin or hemoglobin F (alpha2Agamma2F). During the first year of life, hemoglobin A and A2 gradually replace hemoglobin F, so that the latter constitutes less than 1% of the hemoglobin of adults. Other normal hemoglobin (Gower I, II -alpha2Aepsilon2) are found only in the embryo! All of the normal hemoglobin contain two pairs of polypeptide chains, and to each chain is attached one haeme group.
Abnormal hemoglobin
All of the abnormal hemoglobin is inherited defects. The spontaneous appearance of such mutations is exceedingly rare. There are two types of disorders of hemoglobin. The qualitative disorder is known as haemoglobinopathies (hemoglobin S, C, D, E, O and associated diseases) and the quantitative disorders are know as thalassaemias (Major, intermediate, minor). However, they can be detected by simple laboratory tests. When a person is found to have inherited one abnormal gene he is referred to as heterozygous or having, the trait for abnormality. If he has inherited two abnormal genes the disorder may be referred to as homozygous or doubly heterozygous.
Thalassaemia syndromes
It is now recognized that thalassaemias and related disorders comprise a heterogeneous group of inherited quantitative disorders of hemoglobin synthesis. They are the result of autosomal (nonsex-linked) gene mutations. There are four recognized forms of alpha and beta thalassaemia (depending on the type of globing chain involvement) each with a homozygous and heterozygous expression. Thalassaemia intermedia and thalassaemia major represent clinically severe homozygous state. Thalassaemia minor and thalassaemia minima refer to considerably milder heterozygous form. The four varieties of alpha thalassaemias are alpha thalassaemia-1 trait, alpha thalassaemia-2 trait, HbH disease and Hydrops foetalis (Hb-Bart's disease).
The one common denominator of all the thalassaemia syndromes is a decreased rate of synthesis of one or more hemoglobin polypeptide chains leading to hypo chromic microcytic anemia. [1]. The consequences are precipitation of the free polypeptide chains in red cell corpuscles to form inclusion bodies, red cell membrane damage, shortened red cell survival and the kinetic picture of ineffective erythropoiesis [2, 5,4].
The homozygous beta (Cooley 's anemia) and alpha thalassaemia are evidenced by anemia, medullar, and extramedullary blood formation, splenomegaly, striking changes in the bones, pigmentation of various organs resembling that of haemochromatosis. [5]. the symptoms are so severe that few patients survive adulthood. Due to severe anemia and other complications flying is clearly contra-indicated [1, 5]. In heterozygote’s <beta thalassaemia trait) anemia is the only abnormality seen and this anemia is minimal or nil in some cases. Furthermore, most patients have no symptoms whatsoever and live normal life spans. Treatment is not required for this benign disorder [2, 7].
This usually causes only a slight anemia with hemoglobin levels never going below 9gm/ Dl [7]. The most consistent feature of the hemoglobin pattern of thalassaemia minor is an increase in the proportion of HbA2. Normal value is 2.54 + 0.35%. Value found in carriers is 5.11+ 1.35%. With the exception of some carriers of unstable hemoglobin and HbC and HbS heterozygote and homozygote, an increased level of HbA2 has not been found in any other hemoglobin abnormality. This useful diagnostic feature is lost when there is iron deficiency, but is corrected after iron therapy. The increased synthesis of HbA2 probably is due to increased activity at both delta chain loci. HbF either is not detectable at all or, with rare exceptions, is present in extremely small amounts. Peripheral blood smear shows profound microcytosis and hypochromia with target cells but only minimal or mild anemia. MCV is rarely less than 75 fL; PCV is rarely less than 30 - 33%. MCH is reduced while MCHC may be normal or only slightly reduced. Hb electrophoresis shows HbA2 (3.5 - 7.5%) but some forms may have normal HbA2 or elevated HbF. Other findings are poikilocytosis, basophilic stippling and target cells [2].
Patients with beta thalassaemia may be warned that their blood picture resembles iron deficiency anemia and can be misdiagnosed [1]. Alpha thalassaemia trait findings are hypochromia and microcytosis but no anemia. HbA2 and HbF are normal [2, 5], Comparative values of different Hb levels in beta thalassaemia trait and normal’s are given in Table 5.
| Genotype | Clinical | HbA2 % | HbF% | HbA% | Other Hb |
|---|---|---|---|---|---|
| Delta beta | Normal | 2.5-3.0 | 0 | 96-98 | none |
| Delta thai | Nil | Low | 0 | 100 | 0 |
| Beta thal+ | Thai minor | 3.5-7.5 | 1-5 | >90 | 0 |
| (deltabeta)thal | Thai minor | 2.5-3.0 | 5-20 | <90 | 0 |
| (delta beta)Lepore | Thai minor | 1.2-2.6 | 1.3-14 | present | 6-15% Lepore |
| Casel | Thai minor | 5.6% | 0 | 94.3 | nil |
| CaseJI | Thai minor | 6.79 | 0 | 93.21 | nil |
Diagnosis and D/D of Thalassaemia
The main task was to determine whether it was a thalassaemia syndrome, iron deficiency anemia, haemoglobinopathy or haemoglobinopathy associated with beta thalassaemia. Clinical examination, hematologic studies, hemoglobin electrophoresis, measurement of HbA2 and HbF levels and family studies recognize thalassaemia. However, as mentioned earlier, if beta thalassaemia is associated with iron deficiency, the HbA2 level falls, thereby leading to confusion. Hb electrophoresis will reveal the presence of HbS, HbC, HbD or HbE but does not exclude double heterogeneity for a haemoglobinopathy together with beta thalassaemia. In sideroblastic anemias a double population of red cell corpuscles is likely to be noted. In hereditary persistence of foetal Hb in adult life, the hemoglobin’s distributed uniformly among the erythrocytes. In delta-beta thalassaemia this is distributed unevenly. A rapid screening test based on electronic size distribution curves has been reported to be very helpful in distinguishing thalassaemia trait from both normal and sideropenic disorders. [2, 5]
| Hb% | PCV | MCV | MCII | MCHC | |
|---|---|---|---|---|---|
| Norma l | 13-15 | 42-52/3747 | 80-100 | 27-35 | 31-38 |
| Case-I | 13.4gm | 42 | 84 | 27 | 31.8 |
| Case-II | 11.8 | 37 | 84 | 27 | 32 |
| Waiver | 13(M)/12(F) | 38-54/35-49 | 80-100 | 27-35 | 31-38 |
USA tri-service waiver
Anemia. The aero medical concerns are that anemia reduces tissue oxygenation and can be associated with widespread organ dysfunction, particularly when the hemoglobin concentration falls below 10 gm/dl or the haematocrit is less than 30%. Work capacity and the compensation to conditions of hypoxia are also reduced. The World Health Organization recommends that anemia should be considered to exist when hemoglobin levels fall below 13 gm/dl in males and 12 gm/dl in females. Acceptable values of haematocrits are 40-52% in males and 37-47% in females. Minimum requirement for the average of three haematocrits is between 38.0 and 39.9, or 52.1 and 54.0 for males (35.0-36.9 or 47.1-49 for females). The accepted ranges for a RBC count are 4-7 million/cmm for males and 3.8-5.3 million/cmm for females. Acceptable RBC indices are MCV (80-100), MCH (27-35pg) and MCHC (31-38). If no pathology is detected, the member is fit for flying and no waiver is required. A haematocrit falling below 38.0% for males or 35% for females requires consideration. Waivers have been considered on a case-by-case basis. Any anemia associated with pathology is disqualifying, with waivers considered in light of the underlying diagnosis.
Thalassaemia. The aero medical concern in thalassaemia is that it produces a low-grade anemia that can cause problems at altitude. Splenic enlargement and worsening of the anemia can occur under conditions of stress. The waiver is that aviation personnel must meet the haematocrit standards previously listed. Personnel with thalassaemias causing anemia are not fit for flying. Designated personnel who are heterozygous carriers may be considered for waiver provided there are no other haemoglobinopathies, and if the anemia is limited to a mild, microcytic anemia. Patients who have required splenectomy are disqualified from military flying. Table 6 shows comparative values of Hb% and red cell indices of normal, waiver able range and the two cases in discussion.
Disposal
These two cases bring to light the fact that there might be many undetected carriers of thalassaemia trait on active flying duties in many parts of the world. Once these individuals are identified as carriers of beta thalassaemia, the medical authorities face the dilemma about their fitness for flying duties, particularly fighter flying. Anemia with Hb% below 10gm% certainly calls for unfitness for flying duties unless the exact cause is detected and treated. In the above two cases we have seen that both were asymptomatic barring the laboratory investigation findings (Table 3). Serum iron studies were normal and were differentiated from iron deficiency anemia. Hb% and RBC indices were almost within normal limits and not beyond waiver able range as shown in Table 6. Hypoxia is the only problem we envisage at altitude with this trait, but that is countered by the fact that every fighter aircraft uses oxygen system that starts giving air mix as per requirement. No abnormal reaction was seen during the aviation stresses. Anemia was minimal and individuals remained asymptomatic and free from any other disability/pathology. Moreover, during their entire flying career spanning 18-19 years and consisting of 2500 to 3000 hrs of fighter flying experience, they had not met with any accident or incident which could be attributed towards their thalassaemia trait. Hence, the pilots were considered fit for full fighter flying on type. The disposal also conforms the US tri-service waiver standards [1]. Approving Authority approved the disposal subsequently.
Conclusion
These two cases were detected during a review for other disabilities by way of routine investigation. High incidence of thalassaemia exists in India. The possible chances of their being engaged in aviation cannot be ruled out, as there is no screening of the candidates done at entry. However, we have not confronted any problems of this trait. But the likely hazards of this trait remain. It is a policy decision whether people with this trait will be accepted for flying or not. But the trained manpower should not be wasted. Only caution is that they have to be under continuous observation by AMA and monthly blood hemoglobin estimation should be done to ascertain the level of anemia, if any, and use of oxygen whenever cabin altitude exceeds 10,000ft [6, 7].
References
- Harrison's Principles of Internal Medicine. Vol 673. (15th Ed). McGraw Hill Publication; 2001.
- [Google Scholar]
- Survival of 51Cr-labelled red cells in subjects with thalassaemia trait or G6PD deficiency or both abnormalities. Br J Hematology. 1964;10:171.
- [Google Scholar]
- The Thalassaemia Syndrome (2"d ed). Oxford: Blackwell Scientific Publications; 1972.
- [Google Scholar]
- Hypoxia Tolerance Studies in Aircrew with Sickle Cell Trait: A Case Report. in Indian J of Aviation Medicine. 1977;21(2)
- [Google Scholar]
- Hastings Clinical Aviation Medicine. (3 RI Ed). Castle Connolly Graduate Medical Publishing;
- [Google Scholar]




